

Exzellenzcluster NIM: Nanopartikel transportieren Wirkstoffe
 Privatdozent Dr. Carsten Rudolph
Privatdozent Dr. Carsten Rudolphvom Dr. von Haunerschen Kinderspital am Klinikum
Ein Medikament in die Lungen zu bekommen, klingt vordergründig simpel. Üblicherweise funktioniert das so: Man nimmt einen Arzneistoff, packt ihn in ein Aerosol – eine Art Gemisch aus festen oder flüssigen Schwebeteilchen in einem Gas – steckt das Aerosol in einen geeigneten Inhalator, hält ihn möglichst tief in den Rachen, drückt auf einen Knopf und sprüht und atmet die ausgepressten Teilchen tief ein. Die inhalierten Partikel werden von der eingeatmeten Luft getragen und lagern sich dann in der Lunge ab. „Mit herkömmlichen Medikamenten klappt das alles meist sehr gut“, sagt Privat-Dozent Dr. Carsten Rudolph vom Dr. von Haunerschen Kinderspital am Klinikum der Universität München.
Nicht so, wenn es sich bei dem Arzneistoff um ein Gen handelt – chemisch gesehen also eine Nukleinsäure wie DNA oder RNA, die sich zusammensetzt aus einer fragilen Kette vieler einzelner Bausteine. Im Exzellenzcluster „Nano Initiative Munich“ (NIM) haben Rudolph und seine Kollegen in den vergangenen Jahren Aerosole geschaffen und getestet, mit denen sich auch solche instabile Nukleinsäuren in die Lungen verfrachten lassen. 2012 haben die zuständigen Bundes- und Landesgremien den NIM-Cluster um weitere fünf Jahre verlängert. Darin erforschen Mediziner, Physiker, Chemiker, Pharmazeuten und Computer-Experten der LMU und des Klinikums, der TU München, des Helmholtz Zentrums München und der Universität Augsburg alle möglichen Aspekte von Molekülen bis zu einer Größe von 100 Nanometer (Milliardstel Meter). Zum Vergleich: Ein menschliches Haar ist 100.000 bis 200.000 Nanometer dick.
Die Nanopartikel sind so intelligent, dass sie auch die Hindernisse in den Lungen überwinden.
„Das Cluster hat die Nanotechnologie in München inzwischen sehr gut vernetzt“, sagt Rudolph. Der Mediziner und seine Kollegen denken vor allem an die Therapie der Cystischen Fibrose, der häufigsten Erbkrankheit überhaupt: Jedes 3.000. Baby wird mit einer CF geboren. Letztlich bedingt durch einen Gen-Defekt, legt sich vor allem zäher Schleim über die Lungen, nährt Viren und vor allem Bakterien – was zu einer chronischen Infektion und Entzündung führt, die das Organ nach und nach zerstören. Trotz therapeutischer Fortschritte liegt die mittlere Lebenserwartung bei nur 40 Jahren. Seit Jahren versuchen Wissenschaftler, das defekte Gen durch ein intaktes zu ersetzen. Das Team um Carsten Rudolph hat bereits geeignete RNAs entwickelt, die, einmal im Zielort Lungenzellen angekommen, sicher, effektiv und lange anhaltend wirken und die Funktion des ausgefallenen CF-Gens kompensieren (siehe Jahresbericht 2011).
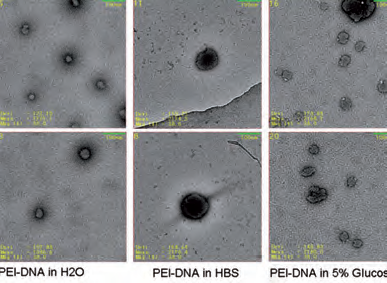 Negativkontrastierung von PEI-DNA-Komplexen unter dem Rasterelektronenmikroskop
Negativkontrastierung von PEI-DNA-Komplexen unter dem Rasterelektronenmikroskop
Bleibt also das Problem des Transports der RNA in die Lungen. „Langkettige Nukleinsäuren werden sehr schnell zerstört, wenn man sie mit großem Druck in herkömmlichen Aerosolen in die Lungen sprüht“, erklärt Carsten Rudolph. Um die Nukleinsäuren vor den entstehenden Scherkräften zu schützen und widerstandsfähiger zu machen, haben sie die Forscher gezielt in spezielle Nanopartikel verpackt. Und zwar so intelligent, dass die Nanopartikel auch die Hindernisse in den Lungen überwinden. Wie ein Schutzschirm liegt über den Lungenzellen eine dünne Flüssigkeitsschicht, die das Organ vor Staubteilchen, schädlichen Stoffen oder Krankheitserregern bewahrt. „Die Schicht behindert natürlich auch ein Nanopartikel, das mit einer RNA beladen ist“, sagt der Pharmazeut, „aber mit der Nanotechnologie haben wir hinreichend kleine und kompakte Konstrukte geschaffen, die die Barriere gut durchdringen können.“
Leicht war das nicht. Immer wieder „hat es irgendwo gedrückt“, erinnert sich Rudolph an die vergangenen fünf Jahre, „aber mit jedem Fehler haben wir die darauf folgenden Systeme optimiert.“ An dieser Stelle beweist sich auch die Stärke eines Exzellenzclusters. Die beteiligten Physiker haben die beim Stofftransport auftretenden Prozesse beschrieben und „uns eher anwendungsorientierten Kollegen wichtige Anstöße gegeben.“ Ein anderes Team um Prof. em. Hildebert Wagner vom Pharmazeutischen Institut der LMU hat jene polymeren Moleküle hergestellt, die mit den Nukleinsäuren beladen werden. Inzwischen ist eine ganze Bibliothek solcherMoleküle entstanden. Sie vereint, dass sie allesamt positiv geladen sind. Damit ziehen sie die unter natürlichen physiologischen Bedingungen negativ geladenen Nukleinsäuren an und verbünden sich mit ihnen. Rudolph spricht von einem „spontanen Selbstanlagerungsprozess“, den die Forscher im Labor allerdings so beeinflussen, dass sich die künstlichen Polymere und ihre RNA-Fracht spontan zu winzigen Kügelchen zusammenknäueln.
Die Technik eigent sich auch für die Therapie anderer Lungenerkrankungen.
Jedes der entstandenen Nanopartikel wurde zunächst in der Zellkultur getestet – und dann im Versuch mit Mäusen. Dafür haben die Wissenschaftler jedes ihrer Polymere mit dem Gen für ein bestimmtes Enzym beladen. Das entsprechende Aerosol haben die Nager dann inhaliert. Nur wenn das Gen-Konstrukt wirklich in den Lungenzellen angekommen war, produzierte die Zellmaschinerie einen Stoff, der die Atemorgane der Mäuse im Licht eines speziellen Mikroskops leuchten ließ. „So haben wir nachgewiesen, dass unsere Nanopartikel die Lungenbarriere überwinden“, erklärt Rudolph.
Ein weiterer wichtiger Punkt: die Sicherheit. Die Nanopartikel dürfen dem Organismus nicht schaden. Dafür spielt das Design der Polymere eine wesentliche Rolle. Sie sind so konstruiert, dass sie nach ihrem Eintritt in die Zellen zügig zerfallen, abgebaut und über die Nieren ausgeschieden werden. Die eingeschleuste RNA hingegen soll möglichst lange in den Lungenzellen verweilen und ihre therapeutische Funktion ausüben – was sie nach jüngsten Erkenntnissen macht.
„Bis jetzt läuft alles nach Plan“, sagt Carsten Rudolph. Auch dank einer Firma, die die Forscher gegründet haben, um ihre Technik schneller an die Patienten zu bringen. In diesem Sinne hat die ethris GmbH jüngst eine Allianz mit einer größeren Pharma- Firma geschlossen. „Wir sind frohen Mutes, dass in den nächsten zwei Jahren erste klinische Studien beginnen.“ Wahrscheinlich mit Patienten, die an der Cystischen Fibrose leiden. Prinzipiell aber eignet sich die Technik auch für die Therapie anderer genetischer Lungenerkrankungen.
Quelle: Jahresbericht 2012 (Text und Bildnachweis)